上一次給大家介紹了表觀遺傳修飾中的DNA甲基化修飾,今天給大家介紹表觀遺傳的另一大修飾組織蛋白甲基化。
核小體是由核心組織蛋白(Histone, H)八聚體(pA × 2、pB × 2、p ×2、p × 2)與纏繞其外周長度為146bpDNA組成的核心顆粒及顆粒之間50bpDNA和一個p構成。
組織蛋白能維持DNA結構、保護遺傳資訊和調控基因表現。組織蛋白胺基末端(N端)結構域伸出核小體,可同其他調節蛋白和DNA發生相互作用。組織蛋白修飾有甲基化、磷酸化、乙酰化、巴豆酰化、泛素化、糖基化、ADP核糖基化等。組織蛋白修飾失衡可導致腫瘤發生發展,且組織蛋白p和p殘基甲基化和乙酰化的喪失已被證實是腫瘤細胞的標誌物。
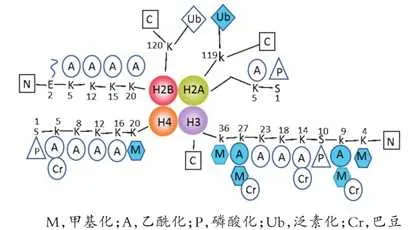
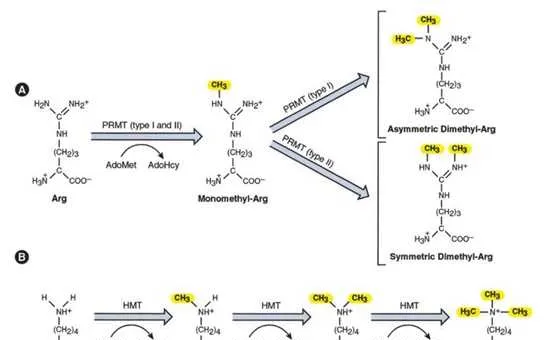
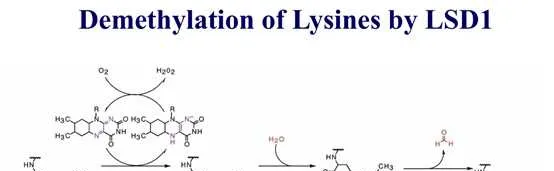
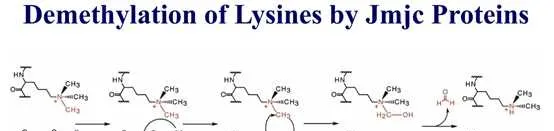
組織蛋白甲基化(histone methylation)是發生在p和p組織蛋白N端賴胺酸(K)或精胺酸(S)殘基上的甲基化。其功能旨在形成和維持異染色質的結構、基因組墨點、DNA修復、X染色質的失活和轉錄等調控方面。組織蛋白甲基化過程主要由組織蛋白甲基轉移酶(histone methyltransferase,HMT)催化,而HMT又可分為組織蛋白賴胺酸甲基轉移酶(histone lysine methyltransferase,HKMT)和組織蛋白精胺酸甲基轉移酶(protein arginine methyltransferase,PRMT)。而組織蛋白去甲基化酶,大致分為LSD(Lysine-specific demethylase)和JMJD(JmjC domain-containing family)兩個家族。LSD1能特異性地去除組織蛋白pK4和pK9的單雙甲基化修飾,而Jmic 家族能去除賴胺酸三甲基化的修飾。p賴胺酸(pK)4、9、27、36、79和p賴胺酸(pK)的20位點可被甲基化,其中組織蛋白pK4和pK9是常見的兩個修飾位點。賴胺酸殘基可以發生單、雙或三甲基化修飾,而精胺酸殘基則只發生單和雙甲基化修飾(對稱或不對稱)。
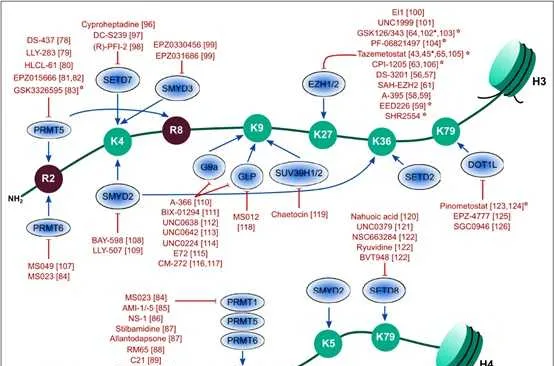
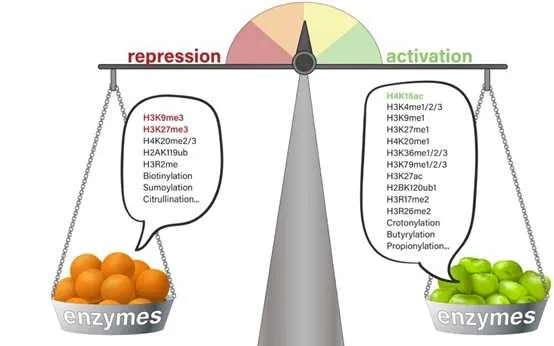
一般來說,組織蛋白p和p不同位點的甲基化以及甲基化數量對基因的轉錄調控具有很大的意義。其中pK9me3,pK27me3以及pK20me2/3介導的是轉錄抑制,而pK4me1/2/3、pK9me1、pK27me1、pK36me1/2/3、pk79me1/2/3介導的則是轉錄啟用。更多的修飾位點所介導的轉錄活性如下圖所示。
1 組織蛋白甲基化修飾酶
賴胺酸可以被六種主要的組織蛋白賴胺酸甲基轉移酶復合物(KMT1-6)單、二或三甲基化。KMT1家族在哺乳動物中至少包含4個成員,包括SUV39p/2、G9a、GLP和SETDB1,並以pK9為甲基化受質。KMT2家族酶存在於大分子復合物中,稱為與Set1相關的蛋白質復合物(COMPASS),並在pK4上進行單甲基、二甲基或三甲基標記。KMT3家族包括NSD1、NSD2 (WHSC1)和NSD3 (WHSC1L1),並主要甲基化pK36。KMT4家族的唯一成員是DOT1L,它實作了pK79的甲基化。KMT5家族包括PR- Set7和SUV4-20p/2,它們分別實作pK20單甲基化和二/三甲基化。KMT6家族包括用於pK27單甲基化、二甲基化和三甲基化的功能冗余酶EZp和EZp。
自賴胺酸去甲基化酶LSD1被發現以來,賴胺酸甲基化一直是一個可逆過程。至少有6個家族的組織蛋白賴胺酸去甲基化酶具有獨特的和重疊的功能。KDM1家族包括LSD1 (KDM1A)和LSD2 (KDM1B),兩者都能使pK4me2/me1脫甲基,但不能使pK4me3脫甲基。此外,LSD1還可以透過將其與CoREST相互作用的抑制復合物轉換為與雄激素受體(AR)相互作用的啟用復合物來參與pK9的去甲基化。與LSD家族不同,賴胺酸去甲基酶的所有其他家族成員都含有Jumonji (JmjC)結構域,由於涉及的化學成分不同,該結構域有潛力去除三甲基標記。JHDM1A (KDM2A)和JHDM1B (KDM2B)屬於KDM2家族,對pK36me2/me1和pK4me3活性較高 。JHDM1A是第一個鑒定出的含有JmjC結構域的去甲基酶。KDM3家族包括KDM3A、KDM3B和JMJD1C,對pK9me2/me1具有去甲基化酶活性。KDM4家族包括KDM4A、KDM4B、KDM4C和KDM4D,對pK9me3/me2和pK36me3/me2具有不同的去甲基化酶活性。KDM5家族包括KDM5A、KDM5B、KDM5C和KDM5D,它們都能對pK4me3/me2進行脫甲基。KDM6家族包括UTX (KDM6A)、JMJD3 (KDM6B)和UTY。UTX和JMJD3對pK27me3/me2有特異性,而Y-連線的類似物UTY催化活性較低。一些KDM被認為是多種癌癥發展的促進因素,因此被認為是潛在的藥物靶點。
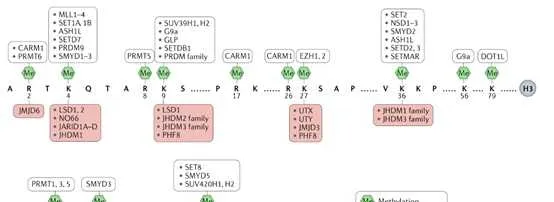
2 組織蛋白賴胺酸不同位點的甲基化修飾
組織蛋白pK4的甲基化是由COMPASS家族中的SET1A、SET1B和MLL1-4在內的強化子和啟動子實作的。COMPASS的不同亞基也被證明可以調控pK4的二甲基和/或三甲基化,包括WDR5、AspL、RbBP5和Dyp30,這些亞基是COMPASS家族所有成員共有的。有趣的是,SET1A的致癌功能已經透過組織蛋白和非組織蛋白受質YAP的甲基化分別與乳癌轉移、肺癌和結直腸癌的腫瘤發生有關。MLL1和MLL2在啟動子和/或Polycomb響應元件(PRE)上實作二甲基化和三甲基化,MLL2也可以在二價基因的啟動子和強化子上實作pK4的甲基化。MLL3和MLL4能夠在強化子處使pK4單甲基化。在急性髓系和淋巴系白血病(AML和ALL)中,MLL1經常透過與其他致癌基因易位而發生突變,在兒童白血病中約占80%,在成人白血病中占5% 10%。嵌合蛋白缺乏MLL1的催化SET結構域,並驅動白血病發生。最近,我們確定了治療MLL重排白血病的策略,透過穩定MLL的野生型拷貝來減弱由MLL融合蛋白及其致癌輔助因子超延伸復合物(SEC)介導的異常轉錄。MLL3和MLL4也在癌癥中均被發現高度突變。組織蛋白pK4me3標記可以幫助招募染色質重塑因子CHD1和BPTF,這兩種染色質重塑因子可以幫助染色質開啟。
由於SET2酶與RNA Pol II的CTD磷酸化形式相關聯,在積極轉錄的基因體內檢測到組織蛋白pK36me3。由ASpL和NSD1-3家族實作的pK36me2的功能不太為人所知。最近,一個潛在的pK4me3和pK36me2之間的串擾在LEDGF的中心發生。LEDGF透過其整合酶結合域直接與Menin和MLL1相互作用,是MLL1依賴的轉錄和白血病轉化所必需的。同時,LEDGF透過其PWWP結構域與二甲基化的pK36結合。LEDGF引起了越來越多的關註,因為研究表明LEDGF在MLL重排白血病中至關重要,而不是在造血系統中,這提高了有效靶向LEDGF而不產生一般副作用的治療潛力。透過使用CP65(一種用於抑制HIV病毒復制的環狀肽)靶向LEDGF,治療mll重組白血病取得了有限的成功,因為LEDGF上的相同結構域結合了HIV整合酶和MLL1。利用蛋白裂解靶向嵌合(PROTAC)技術降解LEDGF可能是一個新的方向。pK36 me3也可以阻止同一組織蛋白尾部PRC2介導的pK27殘基的甲基化。
由DOT1L實作的組織蛋白pK79甲基化標記,位於與活性基因表現相關的組織蛋白的球狀結構域。DOT1L也是唯一一種催化賴胺酸甲基化的酶,它與具有SET結構域甲基轉移酶不同,而pK79的去甲基化酶至今尚未被鑒定出來。DOT1L是在一個名為DotCom的復雜結構中發現的,它與MLL轉移夥伴AF9或其同源物ENL和AF10一起。DOT1L活性還可促進乳癌細胞增殖和轉移。白血病中pK79甲基化異常上調導致了DOT1L抑制劑EPZ- 5676的開發和使用,用於治療MLL重排白血病,目前正在臨床研究中。
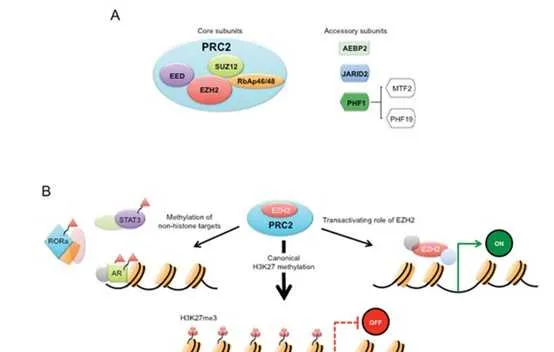
組織蛋白pK9和pK27甲基化是形成不同形式的異染色質所必需的。組織蛋白pK9me3和pK27me3被認為是唯一真正的表觀遺傳標記,因為它們已經定義了在DNA復制後可遺傳的機制。HP1蛋白HP1α (CBX5)、HP1β (CBX1)和HP1γ (CBX3)含有甲基賴胺酸結合的染色體結構域,在異染色質形成中發揮重要作用。SUV39p催化的組織蛋白pK9的甲基化為HP1蛋白創造了一個結合位點,而HP1蛋白又會吸收更多的SUV39p,這一機制有助於異染色質形成的增殖。在pK27me3的形成中,EZp在PRC2復合物中實作pK27甲基化,而EED亞基辨識這種甲基化,並變構性地進一步啟用EZp的SET結構域。與COMPASS家族催化的pK4me1/2/3的獨特分布類似,pK27me1/2/3的分布在整個基因組中是相互排斥的,pK27me3主要位於啟動子(特別是二價基因),pK27me2主要位於基因間區域,pK27me1主要位於轉錄活性基因的基因體中。由於PRC2的EZp和SUZ12亞基對於HP1α的穩定是必需的,因此異染色質標記pK27me3和pK9甲基化可能協同維持染色質上的HP1α,突出pK9me2/3和pK27me3基因沈默途徑之間的關鍵交叉。當EZp在癌細胞中異常表達或突變時,EZp抑制劑常用於防止腫瘤抑制基因的組織蛋白甲基化。
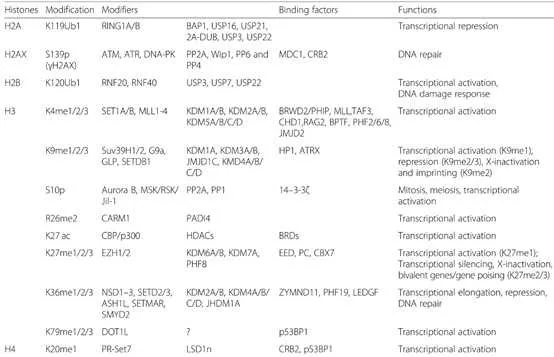
3 組織蛋白甲基化與癌癥
一些證據表明異常組織蛋白甲基化可能在癌癥中發揮作用。最初的研究表明,某些組織蛋白甲基化水平的變化與癌癥復發增加和低生存率相關。盡管這些變化是否具有因果關系尚有待確定,然而,它們可能發展成為潛在的生物標記物,用於藥物發現和診斷或預後。最近的研究提供了越來越多的遺傳學證據,表明組織蛋白甲基化事件在腫瘤發生中起著因果作用。組織蛋白甲基修飾和甲基結合蛋白的突變或表達改變與各種不同癌癥的發病率增加相關。例如,pK27me3甲基轉移酶在一些癌癥中上調,包括前列腺癌、乳癌和淋巴瘤。重要的是,最近發現EZp的啟用點突變與B細胞淋巴瘤相關,這與EZp致癌的觀點一致。與此一致的是,在多種人類癌癥中發現了pK27去甲基化酶UTX的體細胞失活點突變。然而,EZp並不總是作為致癌基因;在骨髓增生症候群中發現了導致EZp甲基轉移酶活性喪失的突變,這表明EZp在該癌癥類別中起著抑癌作用。EZp作為癌基因或抑癌基因的雙重作用突出了癌基因和抑癌基因的背景依賴性,並提出了pK27me3在不同細胞類別中可能具有不同功能的可能性。
D-2-羥基戊二酸酯(D2HG)是一種腫瘤代謝物,可抑制許多去甲基化酶,導致基因組和轉錄組甲基化譜的改變,以及基因表現和基因組拓撲結構的改變。腫瘤發生與編碼異檸檬酸去氫酶1 (IDp)和IDp的基因的特定突變有關,這些突變阻止異檸檬酸轉化為α-酮戊二酸,並促進α-酮戊二酸還原為其結構類似物D2HG。參與組織蛋白去甲基化酶的Jumonji C結構域家族都是鐵依賴雙加氧酶,它們在染色質上的催化活性被高水平的D2HG競爭性地抑制。

4 組織蛋白甲基化靶向治療——組織蛋白甲基化調節劑
在過去的幾年裏,已經開發了大量不同組織蛋白甲基轉移酶的特異性抑制劑。EZp、DOT1L、PRMT1、PRMT5抑制劑目前已進入臨床試驗階段,其中EZp抑制劑的臨床開發最為先進。並且在2020年1月23日,FDA宣布全球首個EZp抑制劑Tazemetostat重磅上市,批準的適應癥為不適用於手術切除的轉移性或晚期的上皮樣肉瘤。
EZpi: EZp是PRC2的催化亞基;其他PRC2亞基包括zeste 12抑制子(SUZ12)、胚胎外胚層發育蛋白(EED)和視網膜母細胞瘤結合蛋白p46(也稱為RBBP7)。EZp透過催化pK27的單甲基化、二甲基化和三甲基化來促進基因沈默。EZp同源物EZp具有較弱的組織蛋白甲基轉移酶活性,但在特定情況下,特別是當EZp水平較低時,仍可能參與pK27甲基化。EZp對B細胞成熟至關重要,是多發性骨髓瘤、濾泡淋巴瘤和彌漫大B細胞淋巴瘤(DLBCL)有前景的治療靶點。約20% - 30%的濾泡性淋巴瘤和生發中心DLBCLs在EZp SET域的特定殘基中存在雜合點突變(Y641, A677, A687),它增強了二甲基化和單甲基化的靶向性,從而導致pK27me3水平的大幅提高。在BRCA1相關蛋白1 (BAP1)突變的惡性間皮瘤和染色質重塑復合物SWI/SNF缺陷的腫瘤中也可以看到對EZp活性的獲得性依賴,例如,在SWI/SNF亞基SMARCB1缺失的惡性橫紋肌腫瘤中。
SWI/SNF核小體重塑復合物可拮抗PRC2介導的基因沈默,並已被證明可從染色質中驅逐Polycomb因子。SWI/SNF失活與EZp突變在一系列癌癥中合成致死。雖然SWI/SNF基因突變被認為透過獲得PRC2功能和沈默腫瘤抑制基因來驅動轉化,但EZp的非催化活性也被認為與腫瘤發生有關。有研究表明,SWI/SNF和PRC2功能的聯合缺失可能導致細胞死亡,這是由於整體轉錄放松,而不是特定PRC2靶點的去抑制。BAP1是一種去泛素化酶,靶向泛素化的pAK119,從而反對Polycomb介導的基因沈默;然而,BAP1遺失後對EZp抑制劑的敏感性似乎是細胞類別特異性的,在間皮瘤中觀察到,但在葡萄膜黑素瘤中未觀察到。PRC2還可以作為腫瘤抑制因子,在骨髓增生異常症候群和慢性骨髓增生性腫瘤中有報道稱EZp反復失活突變。編碼其他核心PRC2成分的基因EED和SUZ12的功能缺失突變也見於T細胞急性淋巴細胞白血病和惡性周圍神經鞘腫瘤。PRC2在不同腫瘤環境中的功能不同被認為反映了特定的細胞轉錄程式和染色質環境在確定PRC2靶向基因方面的關鍵作用,並強調了仔細監測EZp抑制劑治療的患者的必要性。
EZp異常啟用是多種癌癥的一個特征,包括乳癌、去雄前列腺癌、小細胞肺癌(SCLC)和神經母細胞瘤,並已被證實與腫瘤發生和獲得幹細胞樣轉錄程式有關。盡管在某些癌癥中,EZp的上調可能是惡性過程的結果而不是驅動因素139,但臨床前研究表明,在許多這些腫瘤類別中,EZp的缺失或抑制會損害增殖和腫瘤生長。多種EZp抑制劑目前正在一系列癌癥的I/II期臨床試驗中進行評估。令人鼓舞的是,在濾泡性淋巴瘤中,71%的EZp SET域突變啟用的患者對EPZ-6438(他澤美司他)有應答,11%的患者達到完全應答。相比之下,沒有EZp突變的患者只有33%的應答,31%的患者病情進展。
其他抑制劑:其他小分子KMT抑制劑已經被開發為潛在的抗癌治療藥物,包括靶向pK9 KMT的化合物。肺癌細胞系中pK9特異性KMT G9a的表達增加,使用抑制劑BIX-01294可以減少pK9的甲基化。類似地,SETDB1在黑色素瘤和肺癌中經常被放大,mithramycin治療會下調SETDB1以抑制增殖。此外,天然產物毛黴素被鑒定為SUV39H的抑制劑,SUV39H是一種KMT,調節紅細胞和B細胞分化。
PRMTs在癌癥中經常過度表達,因此也成為了有吸重力的抗癌策略靶點。PRMT家族有9個成員(PRMT1-9)將精胺酸甲基化形成甲基精胺酸。I型PRMTs (PRMT1-6和8)催化不對稱二甲基精胺酸的形成,而2型PRMTs (PRMT5和9)催化對稱二甲基精胺酸的形成。組織蛋白精胺酸甲基化標記可以啟用(pR3me2a, pR2me2s, pR17me2a, pR26me2a)或抑制(pR2me2a, pR8me2a, pR8me2s, pR3me2s)基因轉錄,並且在多種癌癥中已經觀察到PRMT功能障礙。在NSCLC中有過表達PRMT1和PRMT4 (CARM1)的報道,在乳癌中PRMT4可以透過解除SWI/SNF的調控來驅動c-Myc通路的表達。在淋巴瘤、白血病、膠質母細胞瘤和前列腺癌中觀察到PRMT5表達增加,據報道這些腫瘤啟用了c-Myc和其他致癌轉錄因子。此外,有報道稱PRMT7可抑制E-cadherin的表達,促進EMT在乳癌中的表達。臨床試驗中的PRMT抑制劑是GSK3326595(原名EPZ015938)和JNJ-64619178,目前正在對實體瘤和非霍奇金淋巴瘤患者進行評估。一些抑制劑已在臨床前研究中顯示出前景,如抑制AML模型中MLL-GAS7或MOZ-TIF2融合轉化功能的PRMT1抑制劑AMI-408,以及在套細胞淋巴瘤異種移植物中顯示抗腫瘤活性的PRMT5抑制劑EPZ015666。
組織蛋白去甲基化酶LSD1 (KDM1A)在幾種癌癥中高表達,並且是造血細胞最終分化所必需的。LSD1通常使pK4me1/2去甲基化,從而抑制轉錄,但當LSD1與雄激素受體相互作用時,其酶活性切換到pK9me1/2,從而刺激轉錄(98)。LSD1也是G9a KMT的受質,在雄激素依賴的基因表現過程中,LSD1的甲基化會刺激CDH的募集。LSD1有一個C端類胺氧化酶結構域,在結構上與單氨氧化酶(MAO)相關。因此,MAO抑制劑tranylcypromine (TCP)可抑制LSD1,但其使用受到任意抗MAO活性的限制。一些更有選擇性的TCP衍生物已經開發出來,其中一些已經進入了臨床試驗。在異種移植中,ORY-1001已被證明可以減少pK4me2和LSD1靶基因的表達,並減少腫瘤的生長。GSK2879552促進AML細胞分化並抑制其增殖。此外,最近有報道稱LSD1抑制劑GSK354和GSK690在體外抑制細胞生長。有趣的是,小分子HDAC抑制劑4SC-202具有抑制LSD1的雙重功能,具有相似的效力,目前正處於晚期血液系統惡性腫瘤患者的臨床試驗中。
雖然有四種靶向LSD1的化合物正在進行臨床試驗,但含有去甲基酶活性的JmjC結構域家族蛋白抑制劑的開發一直比較困難。其中兩種化合物是GSK-J1及其前體藥物GSK-J4,它們能抑制KDM6A (UTX)和KDM6B (JMJD3),但對KDM5A和5B的活性較低。這些蛋白質都與癌癥有關。例如,KDM6A失活突變已在AML、多發性骨髓瘤和膀胱癌中報道,而KDM6B在TALL和轉移性前列腺癌中高表達。此外,在10%的兒童急性巨核細胞白血病中發現KDM5A (JARID1) Nup98融合蛋白。最近,一種KDM5B的強效選擇性抑制劑EPT1013182已被報道在細胞系中具有抗增殖作用,並在異種多發性骨髓瘤模型中抑制生長。