qRT-PCR(即時熒光定量PCR)是RNAi的標準檢測方法,今天小優就以一個siRNA沈默mRNA的案例來展示如何進行計算吧~
RNAi實驗的步驟
本案例采用即時一步RT-PCR分析CDC2基因剔除情況。流程如下:
步驟1: siRNA轉染 - HeLa S3細胞轉染靶向人類CDC2基因的0.5 nM siRNA、陰性siRNA對照(與任何已知的哺乳動物基因沒有同源性的非沈默對照siRNA)。並添加未轉染細胞作為對照。
步驟2: RNA純化 ——48小時孵育後,收集細胞,使用試劑盒純化RNA。
步驟3:使用以上RNA為樣版進行qRT-PCR。使用一步法q RT-PCR試劑盒,(逆轉錄反應和擴增在同一試管中進行)。進行CDC2和GAPDH(一種管家內源性參考基因)的基因特異性定量。

qRT-PCR還需要註意以下內容:
(1)無樣版對照NTC:這是一個無樣版RNA,可用ddpO補足,包含擴增反應的所有成分。NTC可以檢測到qRT-PCR反應組分中可能存在的任何汙染。
(2)內源性內參基因的標準化:內源內參基因是指表達水平在樣本之間不存在差異的基因。將目的基因與內源性內參基因進行比較,可糾正RNA含量的變化和樣本處理中的其他差異。在本實驗中,使用管家基因GAPDH進行歸一化。
(3)來自未轉染細胞的樣版:對未轉染對照的分析顯示,在沒有任何處理的情況下,基因表現水平。
(4)來自陰性siRNA對照的樣版:將轉染陰性siRNA對照的細胞中的靶基因表現與轉染基因特異性siRNA後的基因表現進行比較,計算基因剔除率。
未轉染對照組的基因表現水平應與轉染陰性siRNA對照組觀察到的水平進行比較。二者基因表現應該是相似。這兩個樣本之間的任何基因表現差異都是由非特異性效應引起的。
(5)每個樣本至少重復2次。重復實驗對於解釋樣本間的變化很重要,以確保數據是可靠和穩健的。至少應進行2次重復實驗,重復之間的差異應較低。為了確保變異是可接受的低水平,計算變異系數(%Cv=(標準偏差(重復)/平均CT(重復))x100)。這一點應該始終低於3%。
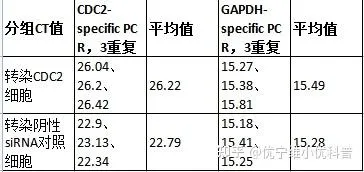
步驟4:結果分析:獲取到可信的CT值,進行計算。本實驗數據如下:
使用∆∆CT方法計算siRNA沈默效率,計算公式如下:
靶基因表現=2-∆∆CT
∆CT (樣本) = CT 靶基因– CT內參基因
∆CT (對照) = CT 靶基因 – CT內參基因
∆∆CT = ∆CT (樣本) – ∆CT (對照)
本實驗數據帶入公式計算: ∆CT(樣本) = 26.22 – 15.49 = 10.73
∆CT (對照) = 22.79 – 15.28 = 7.51
∆∆CT= 10.73 – 7.51 = 3.22
樣本中靶基因表現= 2-3.22 = 0.107,CDC2 siRNA對樣本的沈默效率為:100%-10.7% = 89.3%。
一般來說,下調70%或以上被認為是顯著高的。然而,根據細胞類別、靶基因和下遊檢測的不同,較低的敲低水平可能也就足以用於RNAi研究。
其他的基因調控實驗,如miRNA mimic,cDNA等均可以使用同樣的方法進行計算。
小優推薦:
往期文獻閱讀推薦
#1.siRNA——你不知道的那些小奧秘
#2.為什麽你的siRNA結果不被認可
#3.如何快速啟動siRNA文庫篩選
#4.從培養細胞到qRT-PCR,QIAGEN一步反應試劑盒助力快速基因檢測!
#5.qPCR這件「小」事兒
以上文章可直接進公眾號「優寧維分子生物學」進行搜尋檢視











