【文/觀察者網專欄作者 潘禺】
2016年3月,在觀看了AlphaGo在圍棋這一古老遊戲中擊敗了人類世界冠軍李世石後,DeepMind聯合創始人德米斯·哈薩比斯回想起了自己本科時期的經歷。
他當時玩過一個名為Foldit的遊戲,玩家可以在遊戲中將胺基酸鏈折疊成蛋白質結構,哪怕玩家對生物學一無所知,並不影響他們折疊蛋白質。如果DeepMind能用AI來模仿圍棋大師的直覺,難道不能編寫一個演算法,用AI來模仿Foldit玩家的直覺嗎?
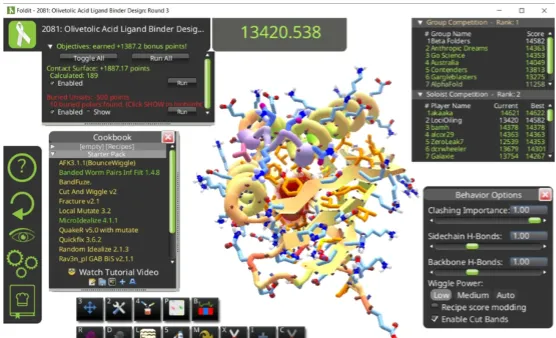
模擬蛋白質折疊的遊戲Foldit的界面
蛋白質折疊問題
蛋白質折疊是一個迷人的問題。
一張紙,在沒有折疊前,不過是壓扁的木漿。當你折疊這張紙,就能產生各種功能。比如折成飛機,那麽紙飛機就能被投擲並滑行,供孩子們娛樂。而如果折成燈籠,就能在中秋節賞玩,表達團圓的美好寓意。
地球上已知的蛋白質,是擁有數億種不同形狀的分子,每一種都執行特定的生物學功能。血紅蛋白和肌紅蛋白在肌肉和身體中運輸氧氣,角蛋白賦予頭發、指甲和皮膚結構,胰島素使葡萄糖進入細胞轉化為能量。這些功能,通常由蛋白質的形狀或結構定義。一串胺基酸分子,在沒有自發折疊成其固有形狀之前,就沒有功能。
一個細胞將稱為胺基酸的小分子串聯成多肽鏈,這就是制造蛋白質的過程。細胞如何選擇胺基酸,取決於DNA提供的底層指令集。多肽鏈一旦組裝好,在極短的時間,千分之一秒內,會彎曲、再彎曲,精確地折疊成蛋白質的最終三維形狀,隨後離開分子裝配線,立即去執行它的生物學工作。
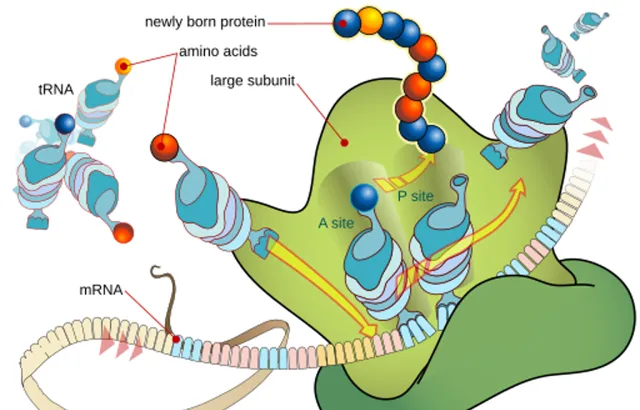
圖為核糖體使用信使RNA樣版制造蛋白質
如果蛋白質執行這種折疊過程出了差錯,錯誤折疊或解纏,就可能導致毒性和細胞死亡。許多疾病,如鐮狀細胞性貧血,就是由錯誤折疊的蛋白質引起的。錯誤折疊的蛋白質聚整合團,是阿爾茨海默病和帕金森病等神經退行性疾病的標誌。
已知的蛋白質結構可以分為四個層次。
一級結構可以理解為一條線性的字串。基本組成單元是一個個的胺基酸,即一個個的字母。常見的胺基酸只有20種,所以一級結構的字串通常只包含20種字母,不包含的6種字母是BJOUXZ。二級結構就是在一級結構的字串的基礎上,肽鏈進行折疊變換,形成一種局部的三維結構。三級結構就是把多個二級結構拼接到一起,折疊成一個完整的蛋白質三維結構。四級結構就是多個三級結構分子組合成一個復合物。

四個不同層次的蛋白質結構
20世紀50年代,生物化學家基斯頓·安芬森的發現使他獲得了諾貝爾獎。他將蛋白質添加到化學溶液中,溶液的破壞導致蛋白質錯誤折疊,但他接下來觀察到,去除化學劑後,蛋白質還是可以自發地重新折疊,恢復其天然的結構。安芬森假設,蛋白質折疊成其原始結構是由蛋白質的胺基酸序列自動完成的,胺基酸序列裏就包含了所需的全部資訊。這就是安芬森教條。
安芬森教條意味著,應該有一種方法可以從胺基酸序列預測蛋白質的形狀,這就是蛋白質折疊問題。
分子生物學中的許多假設被稱為教條(dogma),最著名的是中心法則(The central dogma of molecular biology),遺傳資訊的標準流程是DNA制造RNA,RNA制造蛋白質,中心法則指出,遺傳資訊傳到蛋白質後,不會回流到核酸之中。蛋白質折疊領域還有一個教條,叫萊文塔爾(Levinthal)悖論,說的是一個給定的蛋白質可供選擇的可能構象的數量是天文數碼,即使是一個小蛋白質,也需要比宇宙存在的時間還要多的時間來探索所有可能的構象,可謂「一沙一世界,一花一天堂」。
安芬森教條的例外,則是人類已知的許多疾病。比如朊病毒的構象,就與應有的原生折疊狀態不同。澱粉樣蛋白疾病,如牛海綿狀腦病(瘋牛症)、阿爾茨海默病和帕金森病,都是安芬森教條的例外,原生蛋白錯誤折疊成不同的構象,從而導致致命的澱粉樣蛋白堆積。
回到蛋白質折疊問題,蛋白質組裝的時間這麽短,到底是什麽東西,將蛋白質引向正確的折疊路徑呢?能否從胺基酸序列預測蛋白質的結構?折疊的程式碼和機制是什麽?
為了搞清楚這些問題,至少必須先用實驗確定蛋白質的結構。科學家將蛋白質培育成晶體,用X射線轟擊它們,並測量射線的彎曲,這就是X射線晶體學。20世紀60年代,生物學家馬克斯·佩魯茨和約翰·肯德魯用這種方法確定了血紅蛋白和肌紅蛋白的3D結構,又一項獲得諾貝爾獎的工作產生了。
隨著更多蛋白質結構被發現,科學家們在1971年建立了蛋白質結構的免費檔案庫——蛋白質數據銀行。最初,只包含了七種蛋白質的結構。近50年後,谷歌DeepMind使用它來訓練AlphaFold時,已經包含了超過140000種。
因為方法的繁瑣,為蛋白質銀行添磚加瓦的過程,曾經是非常艱難痛苦的。科學家們先要建立蛋白質電子密度圖,在電子聚集的區域可能包含一個原子。將電子密度圖打印到塑膠片上,一個個堆疊起來,就建立了蛋白質地理的「等高線圖」。然後,科學家們要將地圖轉換為物理模型,將塑膠地圖放入理查茲盒中,這個器材以發明者牛津大學生物物理學家理查茲的名字命名,在理查茲盒內,一定角度的鏡子將地圖反射到工作區,使科學家能準確看到每個原子的相對位置。然後,科學家們就用球和棍子物理構建他們的模型。
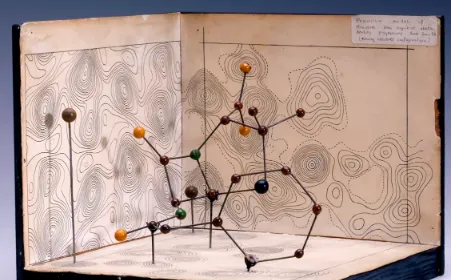
為了研究並模擬磷酸化酶,科學家不得不爬上梯子進入一個特別建造的、有兩層樓高的理查茲盒中。這種蛋白質擁有842個胺基酸,是當時人們研究過的最大的蛋白質。由於進展的煎熬和緩慢,蛋白質銀行成立的20年後,有信心確定而被送出的蛋白質結構也不過七百多種。
實驗主義與計算主義
主張計算方法的科學家,已經厭倦了實驗派的做法,他們希望另辟蹊徑。
正如安芬森的教條,蛋白質的結構應該能從其胺基酸序列中預測出來。計算生物學家編寫電腦演算法,希望可以給程式輸入一串胺基酸,生成正確的蛋白質結構。對計算方法來說,蛋白質的三維結構預測問題,可以看成這樣一個問題,輸入是一個字串,輸出是每個字元(殘基)對應的三個扭轉角ϕ、ψ和ω,看起來簡潔漂亮。註意,這看起來和AI處理的一些經典問題,如序列標註、機器轉譯等問題很像。
計算派的做法是在虛擬世界構建自己的模型,設計自己的演算法,比如假定原子以某種方式粘在一起,蛋白質總是這樣向右或向左折疊,但這些模型逐漸遠離現實。
實驗主義者工作精確但速度慢;計算主義者工作迅速,但與生物物理現實脫節,常常出錯。兩種方法的優點,必須結合起來。實驗派和計算派的科學家,必須牽手合作。
物理學家普朗克有過一句名言:「一個新的科學真理的成功,並不是因為它征服了那些反對者並使他們頓悟,它的成功是因為它的那些反對者最終逝去,而心向新理論的新生代最終成長起來。」
普朗克說的應該是科學理論,是有哲學高度的理論解釋。或許正因為理論還難以建立,在蛋白質生物學的發展歷程中,我們看到的並不是這樣殘酷的規律,而是反對派之間的合作共進。在20世紀90年代,科學家們組成了社區,實驗主義者提供最新的蛋白質胺基酸序列清單,計算主義者則盡其所能,用他們想要的任何方法來預測蛋白質的結構。一個獨立的科學家小組,透過將計算派的模型與實驗確認的結構進行比較,來評估模型。
這個名為CASP的社區,成了解決蛋白質折疊問題各種計算方法的試驗場,最後實際上已經變成了一場競賽。在美國加州的一座老教堂裏,計算主義者可以在會議中談論他們的方法,組織者鼓勵與會者,如果不喜歡他們聽到的內容就在木地板上跺腳。據一位生物學家回憶:「一開始,有很多跺腳,幾乎就像打鼓一樣。」
一些方法的表現比預期好,比如「同源建模」,比較已知蛋白質的結構來推斷未知蛋白質的結構。其他的則完全沒有用。在1998年的比賽中,大衛·貝克用他的演算法羅塞塔(Rosetta)大放異彩,羅塞塔演算法模擬了胺基酸分子間原子的相互作用,以預測它們將如何折疊。盡管還不夠準確,無法實用,但人們看到了計算預測蛋白質結構的曙光。
2008年,貝克建立了一個名為Foldit的免費線上電腦遊戲,也就是本文開頭所說的那個遊戲。在當時,人類玩家模擬蛋白質超過了羅塞塔,但人類的領先優勢不會持續太久。
如果兩個胺基酸一起突變,它們可能有某種聯系,可能在空間上很接近,這一概念被稱為共同前進演化。在清除了統計方法引入的錯誤後,科學家提高了對哪些胺基酸共同前進演化的預測準確度,基於此,羅塞塔演算法能更準確預測蛋白質結構,這可能是深度學習之前推動該領域進步的最大裏程碑之一。但共同前進演化需要大量相似的蛋白質進行比較,而實驗主義者解析蛋白質結構的速度不足以滿足計算主義者的需求。
新玩家上場
2016年,谷歌DeepMind的人工智能團隊以深度學習演算法在圍棋中擊敗了人類冠軍,轟動了世界。
深度學習本身就是電腦科學受到生物學啟發的範例。在大腦皮層中,分子資訊被發送到神經元相互連線的網絡中。神經元有叫作突觸的小臂,它們抓住鄰近神經元發出的分子,這些分子告訴接收神經元要麽發射並傳播訊號,要麽不發射。
將電子位連線起來建立「神經網絡」的想法,早在20世紀50年代就已經在電腦科學中產生。神經網絡中的每個單元是一個節點,可以比作神經元:一個神經元從其他神經元接收資訊,然後計算是否向接下來的神經元發射。在神經網絡中,資訊在多層神經元中傳播,以產生特定的結果,比如影像辨識。神經元層數越多,可以執行的計算就越復雜。
這一靈感正是來自大腦。神經科學發現,我們的大腦會透過逐步抽象的方式來分析眼睛所看到的事物。在AI套用中,輸入數據的傳感器可以是鏡頭、麥克風或者其他測量儀器。而我們人類眼睛中的傳感器又被稱為視錐細胞和視桿細胞,它們會探測那些令其進入激發狀態的光線,得到光線的亮度和顏色。這相當於電腦影像中每一個像素的亮度和顏色。人類的第二層神經元會連線著眼睛的視錐細胞和視桿細胞,一般會衡量相鄰像素之間的相關度,根據上一層神經元的啟用情況來計算。下一層神經元可以在眼睛看到的影像中找出明顯的線條,再下一層,會將線條結合起來,得知影像中的基本物件,比如綿羊的耳朵。再之後的層次,繼而將這些基本物件結合起來,確定更深層次的結構,比如影像中是否存在綿羊。
2010年代初,電腦科學家已經能更好構建神經網絡,允許更多層的可靠訓練。網絡深度從之前的兩三層,躍升到數千層。為了區分過去淺層的做法,人們開始用「深度學習」這個更時髦的名字來稱呼。深度學習改變了人工智能,演算法不僅在影像和聲音的辨識上表現出色,在圍棋這樣的遊戲中也能擊敗人類。近年來,基於深度學習的自然語言處理模型GPT,則在文本生成上又一次震撼了世界。
這裏多說幾句題外話,當前的人工智能革命,還與一種概率論思想——貝葉斯方法(Bayesian methods)有關。貝葉斯方法的核心思想是根據觀測數據更新先驗概率,得到後驗概率分布。貝葉斯方法將不確定性視為概率分布,能夠量化模型的不確定性。在深度學習中,許多問題涉及到對不確定性的建模,例如參數估計、預測的置信度等。用貝葉斯方法,能夠更加靈活地處理這些問題。
一些科學家甚至相信,我們的大腦就是一個能對貝葉斯公式進行各種各樣近似計算的小算盤,也就是貝葉斯大腦,貝葉斯公式很可能在人類認知中處於核心位置。貝葉斯主義者的信念也深刻影響了當前人工智能的發展。總之,「生物學太重要了,不能只留給生物學家」,為了努力理解不同的蛋白質如何折疊,人們不僅要研究生物,還要研究數學、物理、化學、統計學、電腦科學……
百圖生科首席AI科學家宋樂在談到其大模型時就說過:「不單單需要AI人才,也有工程人才的參與,此外還需要一些很了解生物知識、對生物數據分析很有經驗的人才。這種團隊的內部合作不容易,但如果成功也會收效頗豐。」
隨著谷歌DeepMind進入蛋白質結構預測領域,受生物學啟發的深度學習,現在要來解決生物學中的難題了。
AlphaFold的小小震撼
DeepMind的這個專案稱為AlphaFold,來自統計學、結構生物學、計算化學、軟件工程等領域的專家,在DeepMind共同研究蛋白質折疊問題。在學術界,專家們通常相互隔離,各自獨立進行專案,很少有這樣的合作,更沒有谷歌龐大的財務和計算資源支持。2017年,蛋白質數據銀行已經擁有超過140000種結構,DeepMind團隊用這些數據訓練他們的演算法。
其領導者約翰·賈姆珀(John Jumper)正是在物理、化學、生物學和電腦方面有著多樣化的背景。賈姆珀從小自學了編程,本科學習數學和物理,先攻讀凝聚態物理學博士,後來退學在紐約的一家公司用超級電腦從事蛋白質的模擬,透過理解蛋白質的運動和變化,希望更好地理解各種疾病,如肺癌的機制。此後又在芝加哥大學學習理論化學,完成了博士學位。

約翰·賈姆珀(John Jumper)
2018年春天,AlphaFold已經準備好參加CASP,人工智能要與真正的蛋白質科學家競爭了。CASP組織者最終帶來的訊息是,AlphaFold表現得非常好,在預測蛋白質結構方面,比第二名的團隊好大約2.5倍。但這離解決蛋白質折疊問題還很遠。
在賈姆珀的領導下,AlphaFold被更新重建了,DeepMind設計了一種新型的Transformer架構,神經網絡調整了其連線的強度,以建立更準確的蛋白質前進演化和結構數據表示。
AlphaFold2的預測效率和準確性有了巨大提高。DeepMind找了大約50篇發表在【科學】、【自然】和【細胞】等高端期刊上的論文,這些論文都是實驗主義者的辛勤工作成果,描述新的蛋白質結構和功能,將AlphaFold2的預測結果與之對照,可以繼續打磨改進。
在2020年的CASP比賽中,評估員將預測的蛋白質結構與經過驗證的實驗結構進行比較來打分,100分即模型和現實在原子層面上完全匹配。AlphaFold2的大多數結構都達到或超過了90分。大多數情況下,演算法都有效。
DeepMind已經解決了蛋白質折疊問題中的結構預測部份。AlphaFold2能夠準確地根據其胺基酸序列預測蛋白質的結構。對於因疫情封鎖在家,透過Zoom參加CASP會議看到AlphaFold2演示的科學家們來說,這個小小震撼意味著,蛋白質科學的世界已經永遠改變了。
設計蛋白質:逆蛋白質折疊問題
長期以來,實驗生物學家對計算持懷疑態度,AlphaFold2的成功無疑改變了這一點,但如果說「改變一切」,就有些誇大其詞。
AlphaFold2並不等於結構生物學家的失業。
當然,失業總會存在。一些細胞生物學家和生物化學家過去常常與結構生物學家合作,現在他們已經用AlphaFold2來取代。盡管,訓練AlphaFold的數據,是結構生物學家過去用一個個實驗精心確定的。
結構生物學的技術,除了前面說的X射線晶體學,還有冷凍電鏡、NMR 波譜、雙偏振幹涉測量等技術。
而中國公眾可能對冷凍電鏡比較熟悉。這一昂貴的器材(Cryo-EM,冷凍電子顯微鏡),其原理是快速冷凍生物樣品並用電子束轟擊它們。X射線晶體學需要蛋白質結晶,而冷凍電鏡能夠處理非晶態樣品。X射線晶體學在高分辨率原子級結構方面有優勢,而冷凍電鏡在解析大型復合物和動態過程中更為強大。過去十年中,冷凍電鏡發展迅速,成為解析復雜生物大分子結構的重要工具之一。廣為公眾熟知的中國科學家施一公、顏寧等人,都是用冷凍電鏡解析蛋白質結構的專家。

結構生物學家顏寧
如果結構生物學家僅僅研究蛋白質結構,那他們當然失業了。但結構生物學家的目標是發現蛋白質的功能。有了AlphaFold2,他們就有了一個更好的工具,可以在幾分鐘內建立一個假設,而不是等待幾個月甚至幾年透過實驗來確定一個結構。
結構生物學家的角色不僅僅是獲取結構數據,還包括解釋這些數據、設計實驗驗證假設,並理解蛋白質功能和與疾病相關的機制。這個問題就像AIGC會不會讓創作者失業。ChatGPT能告訴你的答案也許準確度已經很高,但未必完美,對AI大模型生成的內容,每一個創作者都還需要仔細甄別、驗證,並理解這些內容的真正意義,用這些內容為自己和社會創造價值。
AlphaFold2的不完美在於,在預測簡單的小型蛋白質結構方面,非常出色,但在預測包含多個部份的蛋白質,動態蛋白質(與其他分子相互作用時,形狀會發生變化)時,準確性較低。有時,蛋白質需要被特定的離子、鹽或金屬包圍才能正確折疊,自然環境會改變蛋白質的形狀,AlphaFold2並不能考慮。
僅僅辨識已知蛋白質的結構和功能是不夠的。
對於新藥研發來說,科學家需要設計那些在自然界中不存在的蛋白質,這就是蛋白質設計,也可以理解為「逆蛋白質折疊問題」。還記得用AI解決蛋白質折疊問題是什麽意思嗎?無非就是向深度學習演算法輸入胺基酸序列,要求其輸出蛋白質結構。這個問題逆過來,就是設計師將一個蛋白質結構輸入演算法,並要求其輸出胺基酸序列。然後,設計師使用那個胺基酸序列在實驗室中構建蛋白質。
宋樂就曾講過,要設計一段有效的蛋白質,「有20個不同的位置,每個位置有20種不同的選擇。這是一個巨大的空間,人的思維很難對這個空間進行整體的篩選或對比,而電腦來做這件事就有巨大優勢。」
Foldit遊戲的建立者貝克,就做了一個專門用於設計的演算法,稱為RoseTTAFold diffusion,Foldit遊戲本身也更新了設計蛋白質的版本。蛋白質設計並非新鮮事物,但深度學習加速了其發展。以前,訓練有素的蛋白質設計師需要花費數周或數月的時間,才能建立新蛋白質的主鏈。現在他們可以在幾天內,甚至一夜之間完成。
AlphaFold3與中國玩家
2022年,谷歌DeepMind釋出了全球已知的2.18億種蛋白質的結構預測,這幾乎就是所有。其競爭對手Meta公司也於當年推出了蛋白質結構預測模型ESMFold。
但AlphaFold2仍有缺陷,比如前文提到的無法考慮環境。細胞內部是復雜的生物學環境,充滿了各種分子——蛋白質、訊號分子、信使RNA、胞器等,蛋白質不是獨立工作,而是不斷與其他分子相互作用,這會改變其自身的形式和功能。將細胞分子的景觀渲染出來,視覺化呈現,你會看到非常壯美的復雜性。

真核細胞的細胞景觀橫截面,渲染圖顯示了其復雜而壯美
AlphaFold2的能力限於預測單一蛋白質結構,而要幫助生物學家理解這個復雜原生環境中的蛋白質,就是這一領域現在的發展方向。2024年春,谷歌DeepMind更新演算法,發表了AlphaFold3的論文,大衛·貝克則推出了RoseTTAFold All-Atom演算法,都致力於能夠預測蛋白質互相結合,或與DNA、RNA和其他小分子結合時的結構。
AlphaFold3能預測分子復合物的結構,比如某種在植物真菌中發現的酶。根據業內專家的分析,目前這些演算法的準確性仍有待改進,不太可能很快帶來新藥。一個重要變化是,AlphaFold2的基礎程式碼是開源的,每個人都可以研究演算法並為自己的專案重建,但谷歌沒有開源AlphaFold3。
中國企業也在加入AI+生命科學的領域。基於AlphaFold2演算法改進,華為昇思MindSpore團隊,采用自己的昇騰計算平台,在2022年4月一度拿下CAMEO這一蛋白質結構預測競賽的第一名,這個比賽每周都會線上更新分數和名次。
2020年創立的百圖生科,則致力於搭建「xTrimo」生命科學大模型,這是一個雄心勃勃的超大規模多模態模型體系,在底座通用模型上,除了蛋白質生成模型,還有多個下遊任務模型共同組成。比如靶點發現,也就是免疫細胞擾動後功能變化預測模型。
當發現了一個疾病靶點後,就要設計一個蛋白質。
如果將疾病相關的靶點想象成一把鎖,設計蛋白質就是配鑰匙,要開啟鎖,鎖齒和鑰匙就要契合,這就需要模型來預測。因此蛋白質生成不僅要預測結構,還要預測蛋白質與靶點的契合度,也就是結合的緊密強弱,然後再對AI生成的許多設計做篩選,將最合適的送去試驗。
xTrimo有多個層次,第一層是對單個蛋白質的建模,第二層是對細胞中蛋白質相互作用的建模,第三層是對細胞本身的建模,第四層則是對細胞系統的建模。因而,這個體系不僅能表征單體蛋白質,還能表征蛋白質相互作用、免疫細胞、免疫系統等多層次生物問題,幫助研究者更快發現新的蛋白質、新的細胞形態,發現新的靶點和藥物設計方向。
為此,百圖生科構建了世界最大的免疫圖譜,包含66億個蛋白,超300億條蛋白互作關系,1億個單細胞,以及超6100萬條免疫互作關系和6000億條泛細胞共現關系。
結語
生命體的高度復雜,還遠不是AI科學家使用的龐大但依然有限的數據量就能揭示的。
蛋白質折疊問題仍未完全解決。AI能辨識出給定胺基酸序列可能的折疊模式,但蛋白質折疊過程中,究竟發生了什麽,其中的資訊依然是黑箱。對於理解整件事發生的過程,AI並不能給出答案,深度學習演算法無法告訴我們基於蛋白質的生命機制和本質,無法告訴我們背後的基本物理原理。如果只有結果,沒有過程,這還是科學嗎?
無論如何,科學確實在前進。70年前,人們還認為蛋白質只是一種凝膠狀物質。但今天,我們看到了蛋白質世界的一個又一個結構。

本文系觀察者網獨家稿件,文章內容純屬作者個人觀點,不代表平台觀點,未經授權,不得轉載,否則將追究法律責任。關註觀察者網微信guanchacn,每日閱讀趣味文章。











