原创 huacishu 图灵基因 今天
收录于话题 前沿分子生物学技术
撰文:huacishu
IF=27.601
推荐度:⭐⭐⭐⭐⭐
亮点:
1、作者概述了如何查看现有的几种单细胞RNA测序方法,并强调了这些方法在关于表达式变化的假设方面的差异。
2、作者还说明了他们的观点如何有助于解决生物学上感兴趣的问题。

美国芝加哥大学Matthew Stephens教授团队在国际知名期刊
Nat Genet
在线发表题为「
Separating measurement and expression models clarifies confusion in single-cell RNA sequencing analysis
」的综述论文。在典型的单细胞RNA测序数据集中,高比例的零导致了广泛但不一致的术语使用,如空白和数据缺失。在这里,作者认为这些术语中的大部分是无用的和令人困惑的,并概述了一些简单的想法来帮助减少困惑。这些想法包括:观察到的单细胞RNA测序计数反映了真实的基因表达水平和测量误差,仔细区分这些将有助于理清思路;方法开发应该从泊松测量模型开始,而不是更复杂的模型,因为它很简单,通常与现有数据一致。本文概述了如何在这个框架内查看现有的几种方法,并强调了这些方法在关于表达式变化的假设方面的差异。作者还说明了他们的观点如何有助于解决生物学上感兴趣的问题。

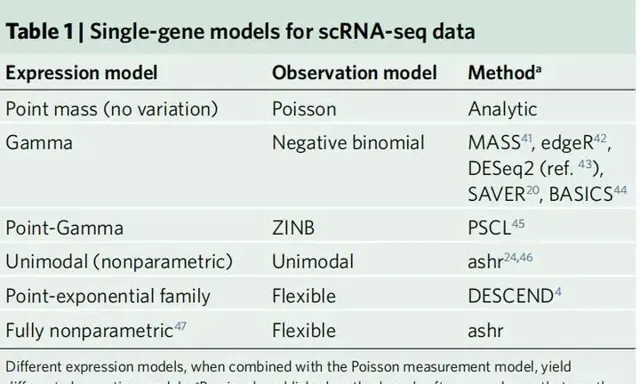
作者强调使用泊松模型特别适用于测量模型而不是观测模型。事实上,许多论文已经证明,泊松观测模型并不能捕获观测到的RNA序列数据的所有变化;因此,通常使用更灵活的观测模型来捕捉额外的变化,例如负二项或零膨胀负二项(ZINB)观测模型。这些观测模型与泊松测量模型并不矛盾;实际上,在本文中作者将解释负二项和ZINB观测模型以及许多其他现有方法是如何通过将泊松测量模型与某些表达式模型相结合而自然产生的。表1给出了单基因表达模型的列表,以及对相应观察模型进行统计推断的一些已发表的方法。
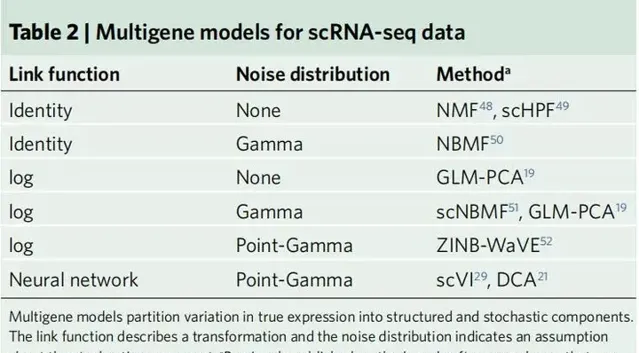
尽管事情不可避免地变得更加复杂,但是类似的想法也适用于多个基因的表达模型。一个多基因表达模型同时描述了细胞内不同基因表达水平和不同细胞间基因表达水平的相关性。描述这些相关性的一种常见且有效的方法是使用低级模型,该模型直观地假设相关性可以通过相对较少的模式(远小于细胞或基因的数量)来捕获。表2中给出了多基因表达模型的列表,以及对相应观察模型实现统计推断的一些已发表的方法。
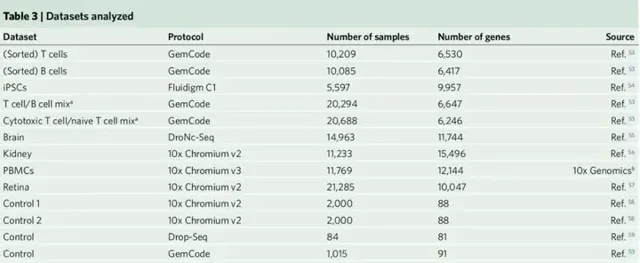
关于scRNA-seq数据是否由负二项观测模型充分建模,或者是否有必要使用ZINB观测模型存在相当大的争论。一些论文得出结论,观察到的scRNA-seq数据表现出多峰表达变异,这表明可能需要一个更复杂的观察模型。在上述框架下,这些问题转化为关于表达式模型:Gamma表达式模型是否足够,或者是否有必要使用更复杂、甚至多模态的表达式模型?由于基因和数据集之间的表达差异可能不同,因此作者从一系列设置中分析数据集,包括分类细胞的同质集合、细胞系和异质组织。同时还建立了分选细胞的硅胶混合物作为高度异质表达模式的阳性对照(表3)。
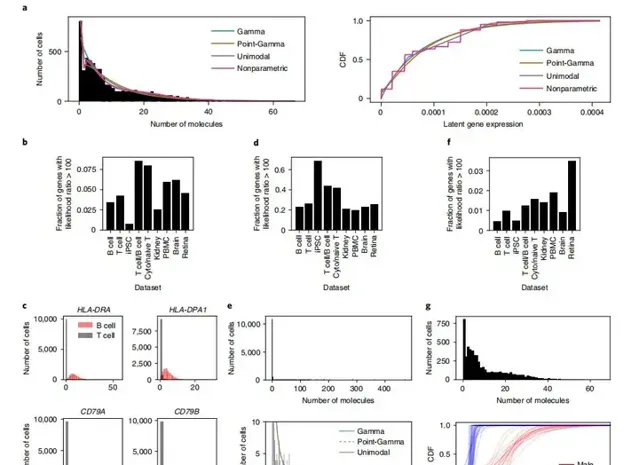
对于每个数据集中的每个基因,作者比较了几种表达模型:伽玛分布、点伽玛分布、非参数单峰分布和完全非参数分布(图1a)。因为这些比较涉及非参数族,所以获取p值并不简单,而且可能不合适,因为将这些模型中的任何一个指定为空表达式模型也是有问题的。因此,作者通过比较每个模型下数据的可能性来比较每个模型的支持度。首先,通过比较伽马表达模型和点伽马模型,作者评估了基因是否显示了由于过多的零而导致的伽马表达模型的证据。在作者研究的所有生物数据集中,只有一小部分基因(0.6–8.6%)显示了支持点伽马模型的有力证据(图1b)。支持点伽马表达模型的有力证据表明,这些基因包括分类的B细胞和T细胞合成混合物中的已知标记基因(图1c),这为这种方法能够找到这种模式提供了一个阳性对照。接下来,通过与非参数单峰表达模型的比较,评估基因是否有证据表明伽玛表达模型存在其他类型的偏离。在这个比较中,更多的基因(20–69%)显示了支持非参数单峰表达模型的有力证据(图1d)。这些结果表明,无论是负二项模型还是ZINB观察模型都不能捕捉到许多基因的表达变异。例如,在外周血单个核细胞(PBMCs)中,PPBP基因不仅表现出许多观察到的零,而且还表现出许多小的非零观察和大的观察的长尾(图1e)。伽马分布和点伽马分布都没有足够的灵活性来同时描述这两个特征,从而解释了非参数单峰模型更好拟合。最后,通过比较单峰表达模型和完全非参数表达模型,评估了数据是否显示多峰表达变异的证据。在这个比较中,很少有基因(0.4–3.5%)显示出完全非参数表达模型的有力证据(图1f),这表明多峰表达变异可能比先前提出的更为罕见。作为阳性对照,RPS4Y1是一个Y染色体连锁基因,由于来自男性和女性供体的诱导多能干细胞(IPSC)中基因表达的不同分布,显示出非参数表达模型优于单峰模型的压倒性证据(图1g)。来自女性供体的细胞被估计具有RPS4Y1的非零表达的一个可能原因是RPS4Y1与其同源物RPS4X之间的编码序列的相关部分是相同的,并且一些读取被错误地映射。
在这里,作者描述了观察到的scRNA-seq计数模型如何被有效地分为两部分:测量模型(描述测量过程引入的变化)和表达模型(描述真实表达水平的变化)。作者认为一个简单的泊松模型是测量模型的合理起点,许多现有的方法可以解释为将泊松测量模型与不同的表达模型相结合。作者解释了这些简单的想法如何帮助澄清关于scRNA-seq数据中零的来源和解释的混淆,并给出了严格的程序来询问细胞间基因表达的变化。如何在scRNA-seq分析中使用这些思想?作者强调,明确区分测量、表达和观察模型有助于减少混淆和误解。特别是,无论是发展统计方法的个人还是分析数据的个人,都应在分析scRNA序列数据时明确对测量误差和表达变异的假设。未来工作的一个重要领域是发展快速和准确的诊断方法,以评估这些假设是否被观测数据所违反,以及分析结果是否对这些假设敏感。
教授介绍

美国芝加哥大学Matthew Stephens教授实验室主要在统计学和遗传学的交叉点上研究各种各样的问题,大部分研究都涉及开发新的统计方法,其中许多方法都有一个非常重要的计算部分。由于数据集越来越大,他们的工作经常涉及现代的「高维统计」方法。经常广泛地使用贝叶斯层次模型来跨数据集或采样单元借用信息。目前的研究领域包括:稀疏性、收缩率和错误发现率,特别是对于复杂的相互关联的数据集;因子分析、降维和大协方差矩阵的估计;聚类方法和归纳;多尺度和小波方法在基因组数据中的应用以及可复制研究与开放科学。
参考文献
Sarkar A, Stephens M. Separating measurement and expression modelsclarifies confusion in single-cell RNA sequencing analysis. Nat Genet.2021;53(6):770-777. doi:10.1038/s41588-021-00873-4











