前言
自噬是一種調節機制,可去除不必要或功能失調的細胞成分,並回收代謝受質。針對腫瘤微環境中的應激訊號,腫瘤細胞和免疫細胞中的自噬途徑發生改變,從而對腫瘤進展、免疫和治療產生不同影響。
最近的研究表明,自噬途徑參與免疫細胞亞群的存活和雕亡、分化、啟用、效應器功能以及向腫瘤的轉運。同時,腫瘤自主自噬可以透過調節免疫反應改變腫瘤生長。此外,透過將免疫檢查點療法與自噬抑制劑相結合,可以消除自噬的促腫瘤作用。因此,自噬在癌癥治療中是一個復雜但很有前景的靶點。
腫瘤細胞的自噬
腫瘤微環境(TME),在癌癥進展、轉移和治療抵抗中起著關鍵作用。在TME中,腫瘤細胞中的自噬可由細胞內和細胞外的應激訊號誘導,包括代謝應激、缺氧、氧化還原應激和免疫訊號。
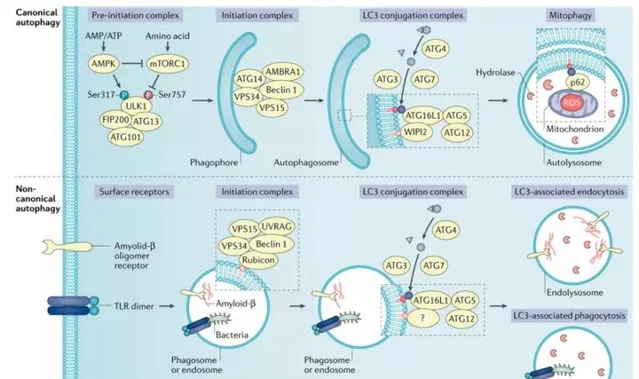
代謝應激
TME攝取的營養不足會影響代謝機制,導致細胞內代謝應激。為了應對代謝應激,腫瘤細胞透過上調營養轉運體和啟用自噬重新構建自己的代謝途徑。
在機制上,5′-AMP活化蛋白激酶(AMPK)和mTOR復合物1(mTORC1)是兩種相反的調節激酶,在營養缺乏的情況下改變自噬誘導。作為一種AMP傳感器,AMPK透過AMP和ATP比率的增加而被啟用,而mTORC1的活性由於胺基酸缺乏而降低。這導致自噬起始前復合物中靶點的磷酸化,從而啟動自噬。
低氧應激
缺氧是實體瘤的特征,缺氧導致粒線體氧化磷酸化受到抑制,AMP與ATP的比率增加,AMPK啟用,缺氧同時也抑制mTOR訊號。此外,缺氧導致啟用轉錄因子4(ATF4)啟用,從而使LC3B和自噬相關蛋白5(ATG5)表達上調,並維持高水平的自噬。
氧化應激
氧化應激反應了自由基和抗氧化劑之間的不平衡。活性氧(ROS)透過氧代謝和其他過程在細胞內產生,過量的活性氧可能增加DNA損傷的風險並促進腫瘤的發生。作為對ROS升高的反應,毛細血管擴張性共濟失調突變(ATM)透過細胞質中的肝激酶B1(LKB1)和AMPK代謝途徑啟用TSC2腫瘤抑制因子,以抑制mTORC1並誘導自噬。氧化應激還可透過NF-κB介導的p62/SQSTM1上調促進自噬。
免疫訊號
免疫訊號可以調節TME中的自噬途徑。損傷相關分子模式(DAMP)和細胞因子是調節自噬反應的主要介質。細胞外DAMP訊號被細胞外或細胞內模式辨識受體感知,如Toll樣受體(TLR),當各種TLR辨識DAMP並啟用下遊訊號時,誘導自噬誘發生。
此外,在果蠅中,細胞因子如TNF和IL-6樣訊號可以啟用自噬,從而促進早期腫瘤生長和侵襲。TGF-β透過SMAD依賴和SMAD非依賴途徑增加BECN1、ATG5和ATG7的轉錄水平,並啟用自噬,在體外可以延緩人肝細胞癌和乳癌細胞的雕亡。
自噬介導的免疫逃避
逃避抗腫瘤免疫反應是各種腫瘤的重要生存策略。最近的證據表明,自噬在腫瘤免疫逃避中起著重要作用。有研究發現,胰腺導管腺癌(PDAC)中MHC I類分子的下調是透過選擇性自噬降解介導的,抑制自噬釋放出強烈的抗腫瘤免疫反應。
另一方面,MDSCs在TME中發揮免疫抑制作用,有研究證明MDSC中的自噬是抑制黑色素瘤抗腫瘤免疫活性的關鍵機制。MDSC免疫細胞中的自噬是降解MHC II類分子的中心,阻止抗腫瘤T細胞的啟動和啟用。
自噬與耐藥性
自噬作為(癌癥)細胞應對威脅性應激源的機制,被認為是癌癥治療中治療抵抗的重要機制。已有證據表明腫瘤細胞對順鉑的耐藥性至少部份是由卵巢癌細胞系中自噬增加介導的。類似的證據表明順鉑、阿黴素和甲氨蝶呤透過抑制骨肉瘤中的自噬來克服化療耐藥性。
有趣的是,例如順鉑和自噬之間的交互作用是一個連續統一體,甚至在體外和體內對某些化療藥物本身不具有耐藥性的細胞中,添加自噬抑制劑,如氯喹(CQ)也可以提高治療效果。這已在腎上腺皮質癌的小鼠模型、結腸癌細胞系和5-氟尿嘧啶以及替莫唑胺誘導的膠質瘤細胞的細胞毒性中得到證實。
除此之外,對基於抗體的治療也進行了類似的結果,例如在曲妥珠單抗耐藥的乳癌細胞中,使用CQ的自噬抑制導致腫瘤的幾乎完全消除。同樣,使用CQ抑制自噬也能夠有效對抗貝伐單抗誘導的結直腸癌細胞自噬,減少體內小鼠腫瘤模型中的腫瘤生長。
自噬在腫瘤治療中的研究現狀
自噬抑制劑分為針對ULK1/ULK2或VPS34的早期抑制劑,如SBI-0206965、3MA和wortmannin,以及針對溶酶體的晚期抑制劑,如CQ、羥基氯喹(HCQ)、巴非黴素A1和莫能菌素,CQ和HCQ透過幹擾溶酶體酸化抑制自噬體降解。然而,在臨床試驗中,HCQ單一療法未能控制晚期胰臟癌患者的腫瘤生長,目前多將自噬抑制與其他癌癥治療相結合,以提高治療效果。
化療
癌癥中的高自噬通量與化療反應降低相關,並與癌癥患者的低生存率相關。臨床前研究表明,抑制自噬可以克服NSCLC、膀胱癌、甲狀腺癌和胰臟癌的化療耐藥性。此外,一些研究的結果表明,自噬抑制可能與MEK–ERK訊號的抑制產生協同作用。
2014年的一項早期II期研究使用HCQ單一療法治療轉移性胰臟癌患者,這些患者以前曾透過其他方法治療,主要終點為兩個月的無進展生存期。結果,不同患者的自噬水平不同程度地降低,但主要終點沒有顯著改善。另一項聯合HCQ、吉西他濱和nab紫杉醇治療晚期或轉移性胰臟癌患者的研究也未能證明延長12個月的總生存期。然而,重要的是,采用HCQ的患者顯示出了顯著且更好的應答率(38.2%對21.1%)。
放療
自噬在保護腫瘤細胞免受放射治療引起的細胞死亡方面起著關鍵作用。在乳癌細胞中,放射線誘導自噬相關基因的表達,伴隨自噬體的積累。在放療的同時短期抑制自噬可增強放療對耐藥癌細胞的細胞毒性。同樣,低氧透過誘導自噬增強A549肺癌細胞的放射抗性。
在膠質母細胞瘤中,放射治療透過增加哺乳動物STE20樣蛋白激酶4(MST4)的表達誘導自噬,該蛋白激酶透過ATG4B磷酸化來刺激自噬。小分子抑制劑NSC185058(靶向ATG4B)與放療聯合使用,可損害膠質母細胞瘤的顱內異種移植生長,並延長治療小鼠的生存期。因此,靶向腫瘤自噬可能增強放射治療的療效。事實上,在癌癥患者的臨床試驗中,已經利用了自噬抑制劑與放射治療相結合。
免疫療法
利用免疫系統是對抗癌癥的重要途徑。抑制自噬可能會損害系統免疫,因為自噬涉及免疫系統發育和效應T細胞的存活和功能。然而,在黑色素瘤和乳癌的臨床前模型中,CQ在短時間內對自噬的系統性抑制並沒失真害T細胞功能。數據表明,免疫系統可能對某種程度的自噬抑制具有耐受性。然而,鑒於自噬可以調節腫瘤免疫反應,靶向自噬可以提高免疫治療的療效,克服免疫治療的耐藥性。
例如,使用抑制劑SB02024或SAR405抑制VPS34激酶活性導致TME中CCL5、CXCL10和IFN-γ水平升高,從而使黑色素瘤和結直腸癌模型中NK細胞和T細胞腫瘤浸潤水平升高。在這些模型中,VPS34抑制也逆轉了抗PD1或抗PD-L1治療的耐藥性。此外,CQ治療可阻斷自噬介導的MHC I類降解,與雙重ICB治療(抗PD1和抗CTLA4抗體)協同作用,在胰臟癌小鼠模型中產生增強的抗腫瘤免疫反應。
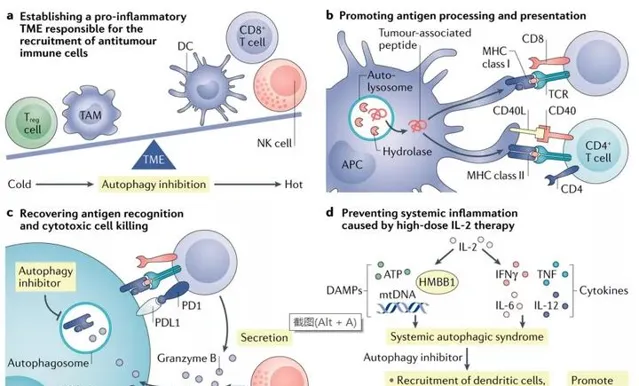
因此,靶向自噬可能增強免疫治療。目前正在進行HCQ聯合免疫治療治療不同型別癌癥患者的臨床試驗。

此外, CAR-T細胞療法在治療血液腫瘤方面取得了臨床成功,但在治療實體癌方面依然療效有限。自噬調節可能為使用CAR-T細胞治療的癌癥患者提供一些益處。眾所周知,TME是實體瘤中CAR-T細胞浸潤和功能的屏障,鑒於自噬抑制可重塑TME並促進Tp型趨化因子的產生,自噬抑制可促進CAR-T細胞向腫瘤的轉運,CAR-T細胞的自噬增強可能支持T細胞在TME中的適應力和存活。此外,腫瘤自噬抑制可能導致抗原表達增加,從而增強CAR-T細胞介導的腫瘤殺傷作用。最後,自噬抑制可能改善細胞因子釋放症候群並為患者帶來臨床益處。總的來說,抑制自噬以提高免疫治療效果的潛力是一個不斷探索的有希望的領域。
小結
自噬是腫瘤生物學、免疫學等多個領域研究的重要機制。鑒於TME中不同細胞中的不同因子可誘導自噬,其誘導和啟用可促進或抑制腫瘤進展。T細胞亞群中的自噬可能在抗腫瘤免疫反應中發揮積極作用,而腫瘤細胞中的功能性自噬可能支持腫瘤抗原呈遞和辨識,在這種情況下,抑制自噬可能有害於抗腫瘤免疫。另一方面,自噬途徑可能與腫瘤細胞存活、腫瘤抗原降解、Tp型趨化因子減少、Treg細胞和MDSC增強有關。因此,以自噬為系統靶向的癌癥治療方法具有挑戰性。
因此,為了在腫瘤免疫治療中靶向自噬途徑,探索主要免疫細胞亞群中自噬的生物學活性至關重要。最近的研究已經開始評估T細胞、巨噬細胞和樹狀細胞的自噬途徑。未來的工作可能會擴充套件到B細胞、NK細胞和NKT細胞。此外,隨著癌癥的進展,自噬途徑會隨著TME中不同刺激的反應而動態改變,必須了解自噬如何同時參與TME中免疫細胞、腫瘤細胞和基質細胞的功能和存活的機制,這樣才能夠最終使癌癥患者獲得臨床益處。
參考文獻:
1.Autophagy in tumour immunity and therapy. NatRev Cancer. 2021 May; 21(5): 281–297.
2. Autophagy in Cancer Therapy-Molecular Mechanisms and Current Clinical Advances. Cancers (Basel). 2021 Nov8;13(21):5575.
更多生物制藥知識盡在微信公眾號"小藥說藥", 歡迎大家關註閱覽!











