原創 驕陽似我 圖靈基因 2021-12-22 07:03
收錄於話題#前沿分子生物學機制
撰文:驕陽似我
IF:41.582
推薦度:⭐⭐⭐⭐⭐
亮點:
1.本文透過在體內穩態和CNS自身免疫炎癥期間對84000多個Tp7細胞的轉錄組和TCR複制型進行命運定位和分析,對組織Tp7細胞的異質性、可塑性和遷移表型進行了表征。器官間和器官內單細胞分析揭示了一個穩態的幹細胞樣TCF1+IL-17+SLAMF6+群體,其透過微生物群運輸到腸道,為IL-23驅動的腦炎性GM-CSF+IFN-γ+CXCR6+T細胞的生成提供了一個現成的儲庫。
2.本文的研究確定了IL-17+非致病性與GM-CSF+和IFN-γ+致病性Tp7人群之間的直接體內關系,並提供了一種穩態腸道Tp7細胞指導腸外自身免疫疾病的機制。
產生白血球介素-17(IL-17)的CD4+T細胞(Tp7細胞)在多種自身免疫疾病中發揮重要作用。然而,最近的研究表明,IL-17和Tp7細胞對自身免疫組織炎癥的驅動作用並不關鍵,而顆粒球-巨噬細胞集落刺激因子(GM-CSF)產生的T細胞(ThGM)被認為是自身免疫組織炎癥的主要誘導因子。此外,Tp7細胞在體內包含高度異質性和可塑性細胞群,產生多種不同的效應和調節性T細胞群。然而,目前尚不清楚組織Tp7細胞是如何將這些不同的訊號整合到一組細胞程式中的,這些細胞程式使它們能夠維持組織內穩態,但卻成為組織炎癥的主要驅動因素。
近期,在
cell
雜誌上發表了一篇名為「
Stem-like intestinal Tp7 cells give rise to pathogenic effector T cells during autoimmunity
」的文章,將單細胞RNA和TCR測序與命運圖研究相結合,以分析84124個組織Tp7細胞,並描述其在穩態和中樞神經系統(CNS)自身免疫期間的異質性、可塑性和遷移。組織Tp7細胞既表現出組織特異性特征,又表現出組織內異質性。在誘導腦脊髓炎(EAE)後,發現了一個幹細胞樣的腸道TCF1+SLAMF6+IL-17+細胞群,該細胞群產生第二個致病性GM-CSF+幹擾素(IFN)-g+CXCR6+Tp7細胞群,該細胞群特異性遷移到中樞神經系統。本文的工作確定了幹細胞樣腸Tp7細胞向致病性Tp7細胞的轉變,並解釋了EAE期間IL-17+與GM-CSF+IFN-γ+致病性T細胞之間的爭議,這一概念可能適用於其他自身免疫和炎癥性疾病。

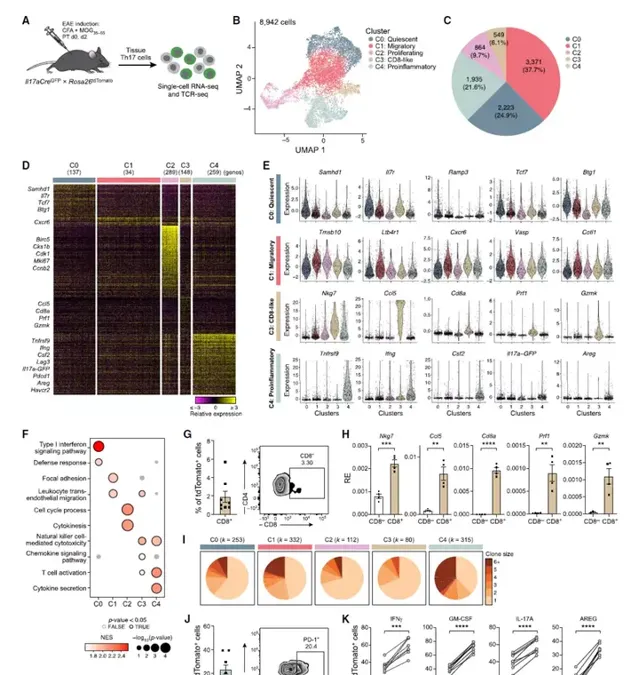
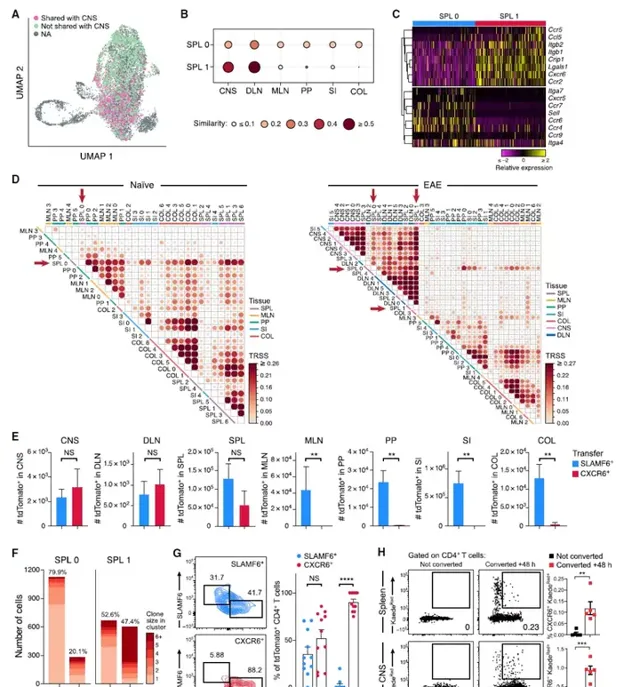
使用scRNA/TCR-seq,分析了46351個在發病時從SPL、MLN、PP、COL、SI、EAE免疫部位腹股溝引流淋巴結(DLN)和CNS分離的tdTomato+組織Tp7細胞。首先關註中樞神經系統浸潤性T細胞。8942個Tp7(TD17+)CNS來源的細胞分為五個簇,透過差異基因標記將其註釋為靜止(簇0)、遷移(簇1)、增殖(簇2)、CD8樣(簇3)和促炎(簇4)。這些細胞在低維空間中從靜止到遷移排列,然後從遷移細胞分裂成增殖細胞、CD8樣細胞和促炎細胞。靜止簇0中的細胞高度表達與T細胞靜止相關的基因(例如Samhd1、Il7r、Tcf7和Btg1),並且每個細胞檢測到的轉錄物數量最低,支持靜止細胞狀態。遷移簇1中的細胞高度表達參與細胞遷移(Ltb4r1、Cxcr6、Vasp)和形態學(Tmsb10、Cotl1)的基因,表明這些細胞最近遷移到中樞神經系統。增殖簇2細胞高度表達細胞周期基因(Birc5、Cks1b、Cdk1、Mki67)。CD8樣細胞(簇3)表達Cd4和CD8+T細胞標記物(Nkg7、Ccl5、Cd8a)和細胞毒性(Prf1、Gzmk)基因。最值得註意的是,發現了一個高度促炎、活化和擴增的細胞亞群(簇4),在中樞神經系統中富集了當前的Tp7細胞。具體而言,促炎簇4中的細胞表達了編碼促炎細胞因子的最高水平的基因,並且每個細胞的轉錄物數量最多。PD-1標記出高度促炎性Tp7細胞,其產生高水平的IL-17A、IFN-γ和GM-CSF細胞因子,這些細胞因子與腦炎性相關。圖1:功能不同的腦源性Tp7細胞亞群的鑒定。
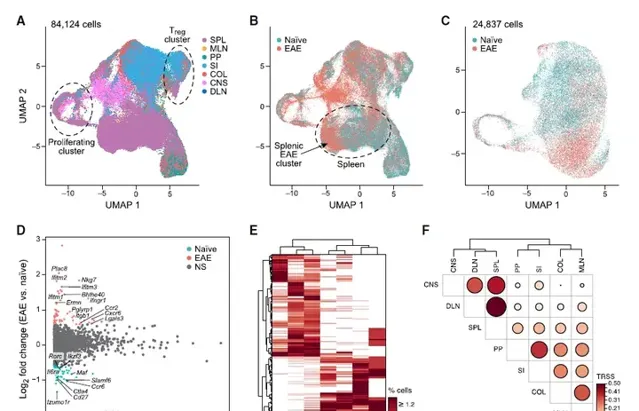
為了研究組織Tp7細胞在EAE誘導中的機制和作用,重點研究了外周組織中的Tp7細胞,並檢測了來自原始和EAE小鼠的所有84124個組織Tp7細胞。結果表明組織Tp7訊號比EAE免疫誘導的表達變化具有更強的鑒別能力。接下來使用EAD小鼠中Tp7細胞的TCR序列來了解所有組織中Tp7細胞與腦源性Tp7群體之間的關系。EAE期間,小鼠之間僅共享少數複制型,而同一小鼠的組織之間共享5%-35%的複制型。組織間的TCR庫相似性分析發現來自DLN、SPL和CNS的細胞之間具有高度複制相似性的Tp7群體,表明這些複制型可能是主要的疾病驅動群體,以及腸組織中具有高度複制相似性的Tp7群體(MLN、PP、SI、COL)。最近的研究強調了「腸-腦軸」的作用,其中腸道Tp7細胞透過未知機制被建議調節CNS自身免疫性炎癥。本文結果提出了一個模型,其中Tp7細胞在DLN中被誘導,然後透過SPL進入CNS。以解決腸道Tp7細胞的潛在作用在EAE疾病中,將腸道Tp7細胞的TCR序列與其他組織Tp7細胞進行比較。有趣的是,在脾臟中發現了一小部份Tp7細胞,它們與MLN、PP、SI和COL中的腸道相關Tp7細胞具有TCR複制型結構。得出結論,SPL中可能存在兩個不同的複制相關Tp7細胞群,一個與DLN和CNS細胞(占SPL細胞的26.2%)共享複制型,另一個較小的SPL細胞群(占SPL細胞的10.6%)。因此假設SPL可能是一個關鍵樞紐,作為傳播腸道Tp7細胞和腦源性Tp7細胞的來源。圖2:EAE期間組織Tp7細胞的單細胞分析。
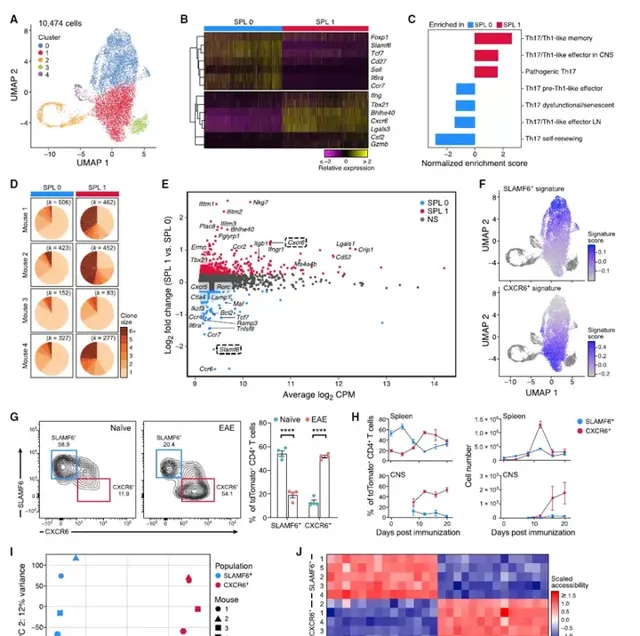
對EAE期間脾臟Tp7細胞異質性的進一步分析表明,大多數細胞(85%)位於兩個簇中的一個,類似於穩態非致病性Tp7細胞(SPL0)或致病性Tp7細胞(SPL1),基於已知基因和特征的表達,致病性樣SPL1細胞大量複制性擴增。在穩態Tp7細胞中上調的基因中,既有先前與非致病性Tp7細胞(Ccr6、Il6ra、Ikzf3、Maf)相關的基因,也有腫瘤和LCMV感染中描述的幹細胞樣CD8+T細胞(Tcf7、Cxcr5)中表達的基因。相反,SPL1細胞表達了和其他人以前與致病性Tp7細胞相關的高水平基因(Ifng、Tbx21、Bhlhe40、Csf2)。將SLAMF6+和CXCR6+鑒定為兩種細胞表面分子,可區分SPL0和SPL1群體,並透過對EAD小鼠脾臟中的SLAMF6+和CXCR6+TdTomatogy+CD4+T細胞進行分類,然後進行大量RNA序列分析,分別驗證其作為SPL0和SPL1細胞的標記,表明它們的相關特征分別在SPL0和SPL1細胞中表達。SLAMF6+細胞在體內平衡期間以高比例存在於脾臟中,在EAE中減少,而CXCR6+細胞在EAE疾病期間在脾臟和CNS中顯著增加,在脾臟中出現臨時峰值,隨後在CNS中穩步上升。值得註意的是,SLAMF6+和CXCR6+標記也在DSS誘導的結腸炎小鼠的tdTomato+CD4+T細胞的scRNA-seq圖譜中標記了不同的細胞群,這表明對其他炎癥條件可能具有更廣泛的意義。脾臟SLAMF6+和CXCR6+群體具有不同的染色質可及性景觀,具有17449個差異可及區域,支持它們的獨立特性。在Slamf6+人群中,參與穩態和幹細胞樣Tp7細胞表型(Il6ra、Ccr7、Ccr6、Slamf6、Tcf7、Cxcr5)的基因座是可特異性存取的,而參與Tp7致病性的基因座(Ifngr1、Nkg7、Tbx21、Cxcr6、Bhlhe40、Csf2、Ifng)在Cxcr6+人群中是可特異性存取的。因此,鑒定了兩個具有不同表達和染色質可及性特征的脾臟Tp7細胞群,它們類似於穩態幹細胞樣細胞群和致病性Tp7細胞群,分別由表面標記物SLAMF6和CXCR6標記。圖3:脾臟內穩態和致病性Tp7群體的發現。
基於對兩個複制相關Tp7細胞群的發現,假設SPL0(穩態)和SPL1(致病)細胞分別代表脾臟中的腸道相關和腦源性Tp7細胞群。事實上,在所有組織中,SPL0細胞顯示出與Tp7細胞相似的TCR序列,而SPL1細胞幾乎完全與DLN和CNS中的Tp7細胞共享TCR。SPL0細胞高度表達參與免疫細胞在淋巴組織(Cxcr5、Ccr7、Sell)和腸道(Ccr9、Itga4)中遷移和滯留的基因,而SPL1細胞表達參與免疫細胞運輸到炎癥部位的基因(Ccr5、Itgb1、Crip1、Cxcr6、Ccr2)。穩態和EAE時所有組織簇的TCR相似性分析揭示了複制型共享的模式。具體而言,在體內平衡時,來自同一組織的細胞簇共享複制型,表明每個組織內的高複制性(和可塑性),並且TCR特異性是決定組織Tp7細胞歸巢和擴增的主要因素。穩態SLAMF6+SPL0細胞與所有組織中的簇具有高度的複制相似性。值得註意的是,在EAE中,只有SPL0細胞與這兩個群體共享大量TCR序列,而SPL1細胞與SPL、DLN和CNS中的所有簇顯示出密切的TCR共享。SPL1與所有CNS簇顯示出強烈的TCR共享,表明SPL1與CNS中的所有Tp7細胞表型相關。值得註意的是,一個小的SI簇(SI5)與疾病驅動生態系和高度表達的遷移基因分組。為了驗證SPL0和SPL1細胞在EAE期間在體內具有不同的遷移行為,將從啟用EAE小鼠獲得的SLAMF6+和CXCR6+SPL細胞分類並過繼轉移到具有同源標記的小鼠中,該小鼠在轉移前7天免疫EAE並收獲轉移後7天(EAE免疫後14天)每個組織中的細胞。CXCR6+轉移群體僅在CNS、DLN和SPL中發現,而SLAMF6+群體存在於包括腸組織在內的所有組織中,證實了EAE期間SPL0和SPL1在體內的不同遷移特征。基於先前的研究表明腸道Tp7細胞可以影響EAE的發展,假設幹細胞樣的腸道TCF1+SLAMF6+SPL0細胞可以產生致病性PL1細胞,並在EAE期間補充這些細胞。為了驗證假設,首先分析了SPL0和SPL1細胞之間的TCR序列共享程度,發現它們之間存在大量複制共享,覆蓋了20%的SPL0細胞和50%的SPL1細胞。SPL0細胞跨越不同的TCR基因庫,擴增的複制較少,這與不同因子誘導的穩態群體相一致,而SPL1的基因庫多樣性較低,但擴增較多。與SPL0細胞相比,共享複制型在SPL1細胞中高度擴增。為了測試SPL0到SPL1細胞的體內轉化,將來自EAE小鼠的SLAMF6+或CXCR6+細胞轉移到具有先天標記的免疫受體小鼠中。移植SLAMF6+供體細胞後,恢復了SLAMF6+和CXCR6+細胞,證實了SLAMF6+細胞在體內產生CXCR6+細胞的能力。相反,在轉移CXCR6+供體細胞後,僅在受體脾臟中恢復CXCR6+細胞,但幾乎沒有SLAMF6+細胞,這表明CXCR6+細胞表現出末端效應細胞表型。最後,透過使用普遍表達光轉換熒光蛋白的Kaede小鼠進行光轉換實驗,直接驗證了脾臟和中樞神經系統中腸細胞向XCR6+細胞的遷移和轉化。在免疫小鼠腸道細胞光轉換後,在脾臟和CNS中檢測到光轉換的CXCR6+CD4+T細胞,這些細胞表達IFN-γ和GM-CSF,證實腸道細胞是致病性CXCR6+細胞的儲庫,這些細胞遷移到CNS以引起神經炎癥。圖4:穩態和致病性脾臟Tp7群體在體內表現出不同的遷移行為。
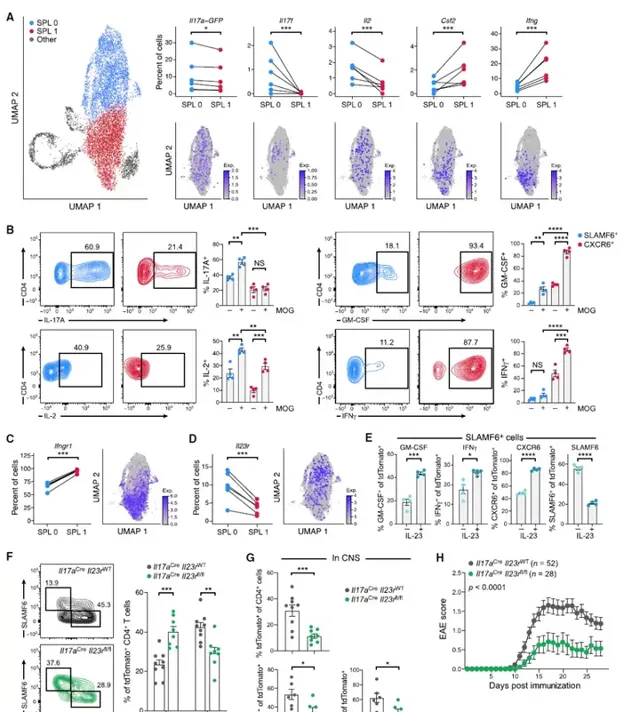
目前尚不清楚產生IL-17的Tp7細胞或產生GM-CSF和IFN-γ的T細胞的獨特亞群是否是導致自身免疫組織炎癥的關鍵致病細胞。為了闡明這個問題,檢測了穩態SLAMF6+和致病性CXCR6+細胞的scRNA序列中編碼關鍵細胞因子的基因的表達模式。有趣的是,SPL0細胞表達Il17a、Il17f和Il2的比例明顯較高,而SPL1細胞表達Csf2和Ifng的比例較高。這種細胞因子基因表現被MOG召回試驗證實為抗原特異性,因為SLAMF6+細胞在用MOG肽體外刺激後顯示出IL-17A和IL-2陽性率增加,而CXCR6+細胞在肽刺激後顯示出GM-CSF和IFN-γ陽性率和表達水平顯著增加。這表明產生IL-17的Tp7細胞和產生GM-CSF-和IFN-γ的T細胞在EAE的誘導中都有作用,因此穩態IL-17+群體產生GM-CSF+IFN-γ+T細胞。此外,表達IFNGR1的促炎性CXCR6+SPL1細胞比例較高,表明IFN-γ介導的自分泌正反饋環在擴充套件或維持SPL1表型方面起作用。
IL-23訊號在自身免疫發病機制中起著關鍵作用,是致病性T細胞生成所必需的,但其驅動致病性T細胞生成的機制尚不清楚。有趣的是,IL-23受體(IL-23R)在SPL0中的表達比例高於SPL1細胞,而在CXCR6+和SLAMF6+細胞中,IL-23訊號在SPL1細胞中的表達更高,在IL-23R訊號下遊的基因位點上的染色質可及性更高,提示CXCR6+細胞中有更大的IL-23R訊號傳導。假設IL-23R訊號可能介導SLAMF6+向CXCR6+細胞的轉化。為了驗證假設,在體外將SLAMF6+細胞與脾細胞共同培養,並用IL-23處理它們。在IL-23存在的情況下,SLAMF6+細胞的比例降低,而GM-CSF+、IFN-γ+和CXCR6+細胞的比例增加,這表明IL-23R訊號介導了穩態幹細胞樣Tp7細胞向致病性CXCR6+細胞的轉化。最後,為了表征IL-23R訊號對體內致病性CXCR6+細胞生成的影響,構建了IL-23R條件敲除小鼠並在Il17aCreIl23rfl/fl小鼠中誘導EAE,其中IL-23R在Tp7細胞中被特異性刪除。與野生型同窩對照組相比,發現脾臟中SLAMF6+細胞的頻率增加,CXCR6+細胞顯著減少,而且,中樞神經系統中的TDT+CD4+T細胞較少,表達GM-CSF和IFN-γ的細胞較少,並且小鼠對EAE的誘導具有抵抗力。Il17aCreIl23rfl/fl CXCR6+細胞的大量RNA序列顯示,在沒有IL-23R訊號的CXCR6+群體中,有大量表達變化。總之,這些數據表明IL-23R訊號介導SLAMF6+細胞產生致病性CXCR6+細胞。圖5:IL-23R訊號正在驅動從IL-17+SLAMF6+細胞生成GM-CSF+IFN-γ+CXCR6+細胞。
本文的研究提供了可能適用於各種自身免疫環境的機制性見解,針對幹細胞樣腸道Tp7人群和IL-23R訊號可以更好地控制許多人類自身免疫疾病的慢性和復發。但存在一定的局限,
本研究中的TCR分析統計數據,包括複制擴充套件和複制相似性評分,即使在標準化後,也可能對細胞數量有剩余依賴性。關於研究設計,研究集中於Tp7譜系,包括所有曾經表達Il17a的CD4+T細胞,因此不考慮其他可能的T細胞譜系.在自身免疫性疾病中發揮重要作用,包括Tp細胞和Treg細胞。
教授介紹:

Vijay K. Kuchroo
Vijay Kuchroo博士是哈佛醫學院Samuel L. Wasserstrom神經病學教授,布萊根婦女醫院的高級科學家,以及波士頓布裏格姆研究所感染與免疫中心的聯合主任。Vijay Kuchroo還是Broad研究所的研究所成員,也是Klarman細胞觀測站計畫和食物過敏科學倡議(FASI)計畫的高級研究員。他是哈佛醫學院和布萊根婦女醫院恒大免疫疾病中心的創始主任。他的主要研究興趣包括自身免疫性疾病- 特別是共刺激的作用 - 實驗性自身免疫性腦脊髓炎和多發性硬化癥的遺傳基礎,以及調節誘導T細胞耐受性和功能障礙的細胞表面分子和調節因子。他的實驗室已經制造了幾只基因改造小鼠,作為人類多發性硬化癥的動物模型。他的實驗室還首先描述了TIM基因家族,並將Tim-3鑒定為T細胞上表達的抑制受體,現在正被用於癌癥免疫治療。他首先描述了一種高致病性Tp7細胞的發展,這種細胞已被證明可以在人類中誘導多種不同的自身免疫性疾病。由Kuchroo博士撰寫的一篇描述Tp7發展的論文是免疫學中參照次數最高的論文之一。
參考文獻:
Schnell et al., Stem-like intestinal Tp7 cellsgive rise to pathogenic effector T cells during autoimmunity,Cell (2021),https://
doi.org/10.1016/j.cell.
2021.11.018











